Nelson Chapter 501-1 Ewing Sarcoma
引用 (16)
 2020/4/8 11:02:04
2020/4/8 11:02:04
 4266
4266
Nelson501.1 Malignant Tumors of BonebyCarola A.S. Arndt
EWING SARCOMA (ES) (20170911 Summary)
Epidemiology
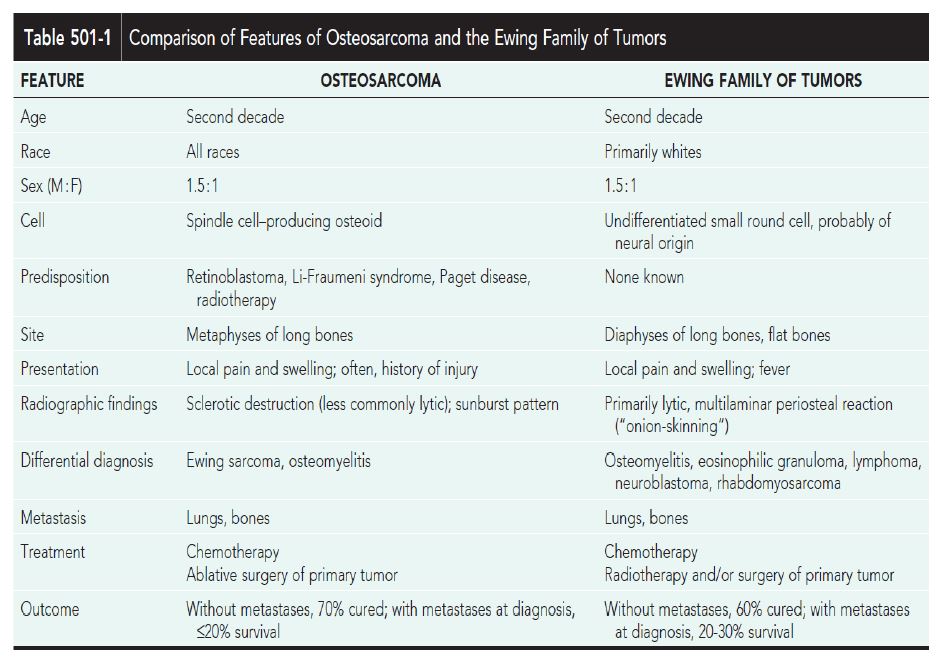
1. incidence: in USA 2.1 cases per 1million children.
2. an undifferentiated sarcoma of bone and softtissue.
3. family of tumors: a group of small, round cell, undifferentiated tumors thought to be of neural crestorigin that carry the same chromosomal translocation, includes Ewing sarcoma &peripheral primitive neuroectodermal tumor (PNET).
4. Treatment protocolsfor these tumors are the same.
5. Anatomic sites of tumors arising in bone: extremities and the central axis (pelvis, spine, andchest wall); Primary tumors arising in the chest wall: as Askin tumors.
Pathogenesis
1. Immunohistochemical staining assists to differentiate it from small, round, blue cell tumors suchas lymphoma, rhabdomyosarcoma, and neuroblastoma.
2. Histochemicalstains may react positively with certain neural markers on tumorcells (neuron-specific enolase (NSE) and S-100), especially in peripheralPNET.
3. Muscle markers(e.g., desmin, actin) are absent.
4. MIC-2 (CD99) staining isusually positive.
5. T(11;22), or avariant thereof is found in most of the Ewing sarcoma family.
6. Analysis for the translocation by FISH (fluorescent in situhybridization) or PCR analysis for the chimericfusion gene products EWS/FLI1 or EWS/ERG is utilized routinely indiagnosis.
Clinical Manifestations
1. Symptoms: are similar to osteosarcoma; pain, swelling, limitation of motion, and tenderness over the involvedbone or soft tissue
2. Huge chest wall primary tumors: respiratory distress
3. Para-spinal or vertebral primary tumors: cord compression
4. Associatedwith systemic manifestations: fever and weight loss; patientsmay have undergone treatment for a presumptive diagnosis of osteomyelitisor a FUO
5. Delayin diagnosis when their pain or swelling is attributed to a sports injury.
Diagnosis
1. S/S pain and swelling, with or without systemic symptoms.
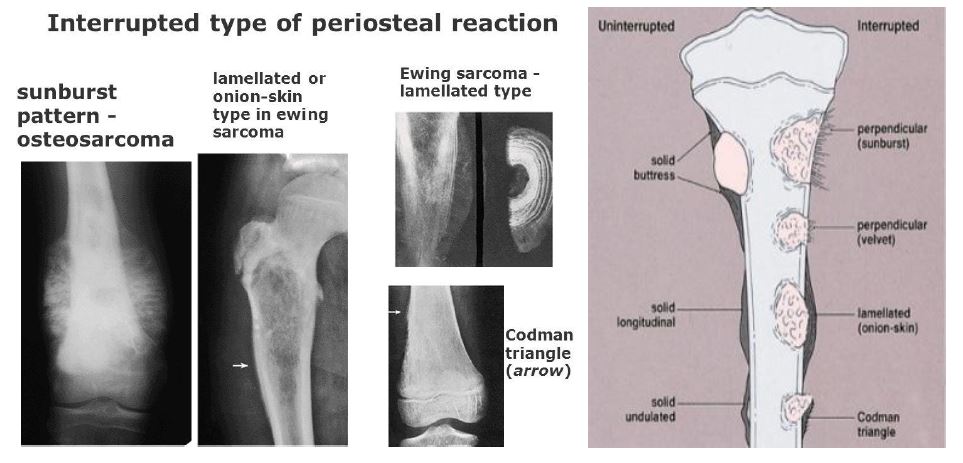
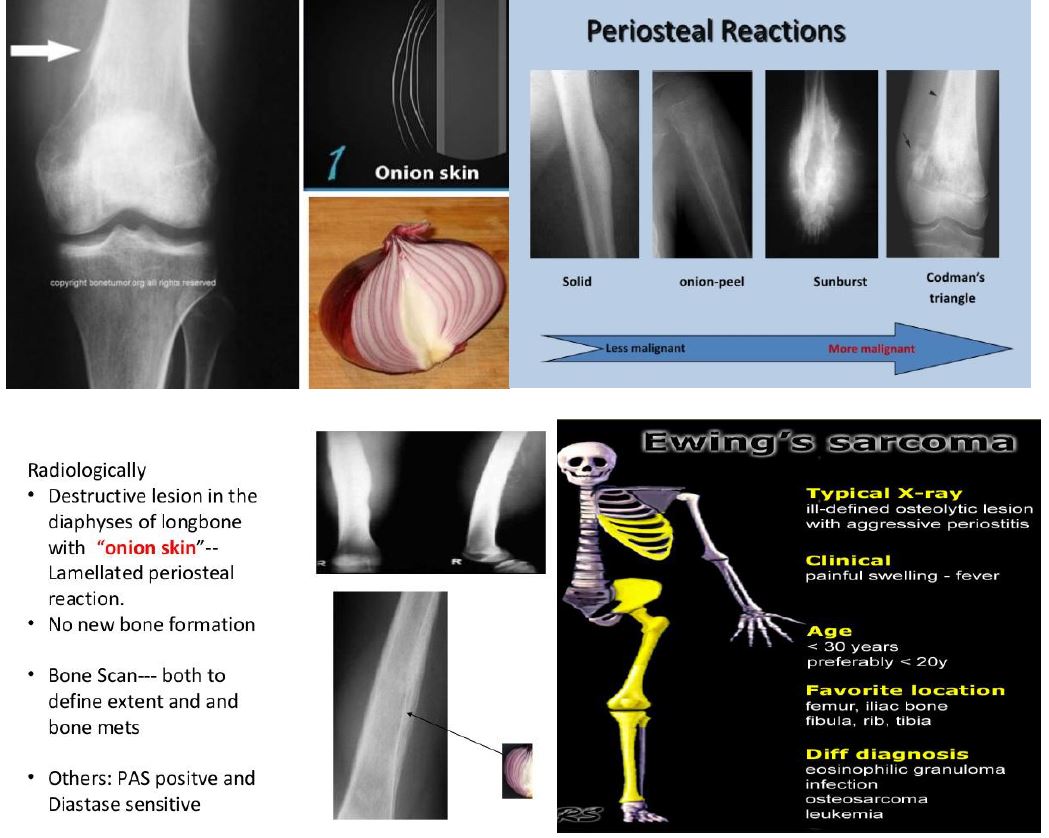
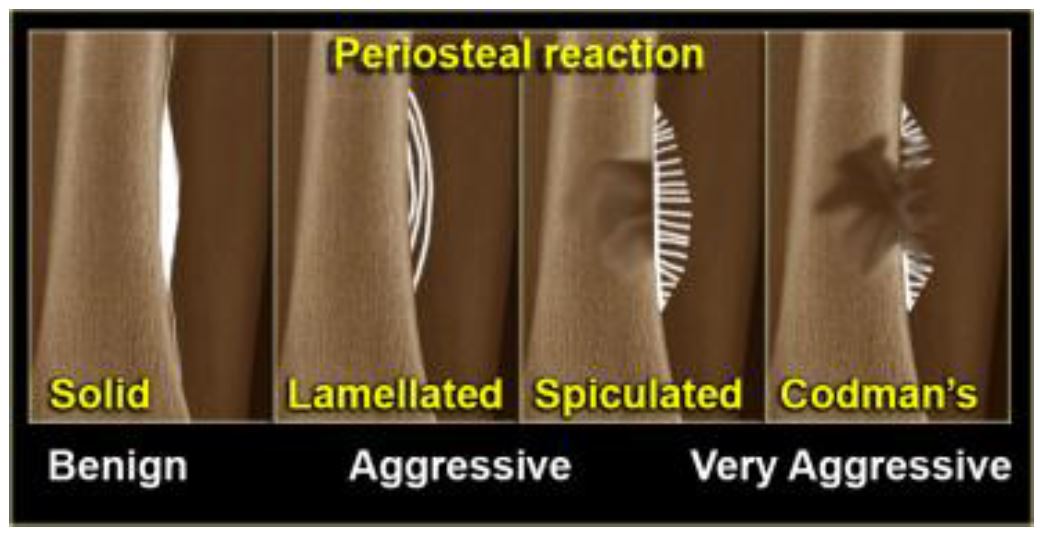
2. Images: a radiographic appearance of a primarily lytic bone lesion with periosteal reaction, the characteristic onion-skinning.
3. A large, associated, soft tissue mass often is visualized on MRI or CT.
4. D.D: osteosarcoma,osteomyelitis, Langerhans cell histiocytosis, primary lymphoma ofbone, metastatic neuroblastoma, or rhabdomyosarcoma in the case ofa pure soft tissue lesion.
5. Evaluations for metastatic disease includes: CT of the chest, radionuclidebone scan, and bone marrow aspiration and biopsy specimens from at least 2 sites.
6. MRI: to determine the exact extension of the softtissue and bony mass and the proximity of tumor to neurovascularstructures.
7. Fluorodeoxyglucose positron emission tomography scanning is being incorporated to enhance staging and to evaluate responseto therapy.
8. CT-guided biopsy of the lesionoften provides diagnostic tissue.
Treatment
1. Standardchemotherapy (USA) for nonmetastatic Ewing sarcoma includes vincristine,doxorubicin, cyclophosphamide, etoposide, and ifosfamide.
2. C/T causes dramatic shrinkage of the soft tissue mass andrapid, significant pain relief.
3. Current studies are evaluatingthe addition of topoisomerase inhibitors to standard chemotherapy, and plans are under way to investigate the role of insulin-like growthfactor receptor inhibitors for certain groups of patients.
4. BMT withmyeloablativeC/T is superior to chemotherapy with lungirradiation for patients with pulmonary metastases.
5. Ewing sarcoma isconsidered a radiosensitive tumor, and local control may be achievedwith irradiation or surgery.
6. R/T is associated with a riskof radiation-induced second malignancies, especially osteosarcoma, aswell as failure of bone growth.
7. Surgical resection, if possible, to achieve local control. Chemotherapy should be resumed as soon as possibleafter surgery.
Prognosis
1. Patients with small, nonmetastatic, distally located extremity tumorshave the best prognosis, with a cure rate of up to 75%.
2. Patients with pelvic tumors have, until recently, had a much worse outcome.
3. Patientswith metastatic disease at diagnosis, especially bone or bone marrowmetastases, have a poor prognosis, with <30% surviving long term.
4. Long-term follow-up of patients is important, even as long as 10 yr after initial diagnosis.